A 75-Year-Old Woman with Increasing Proximal Weakness Treated for Concurrent Idiopathic Inflammatory Myopathy and Myasthenia Gravis
From Grand Rounds from HSS: Management of Complex Cases | Volume 13, Issue 3
Case Report
A 75-year-old previously healthy woman was hospitalized after 3 weeks of rapidly worsening proximal weakness. Physical examination demonstrated marked weakness in neck extension and bilateral shoulder abduction, elbow flexion and extension, and hip flexion. There was no rash or synovitis. Initial laboratory workup was significant for elevated levels of creatine kinase (1,118 IU/L) and C-reactive protein (2.1 mg/dL) and reduced level of hemoglobin (9.3 g/dL). Magnetic resonance imaging (MRI) of the thighs with and without contrast showed extensive bilateral muscle edema involving multiple compartments. Imaging of the chest, abdomen, and pelvis showed no evidence of malignancy, and a transthoracic echocardiogram was unremarkable. Electromyography (EMG) was consistent with diffuse myopathy.
The patient was started on high-dose intravenous methylprednisolone for suspected idiopathic inflammatory myopathy (IIM), with little change in proximal strength, but a decline in creatine kinase level was noted. Two days later, the patient noted new dysphagia, muffled voice, and worsening dyspnea, with a rapidly escalating supplemental oxygen requirement over a matter of hours requiring initiation of bilevel positive airway pressure (BiPAP). She received pulse-dose steroids and was started on plasma exchange (PLEX).
Further laboratory workup showed strongly positive acetylcholine receptor binding, modulating, and blocking antibodies, consistent with myasthenia gravis (MG). An extended myositis autoantibody panel was negative. Thigh muscle biopsy showed marked infiltration of T cells (predominantly CD8+) and histiocytes/monocytes, including formation of multinucleated giant cells (Figures 1 and 2). Microorganism stains were negative, and there was no immunohistochemical evidence of histiocytosis or other neoplastic process.
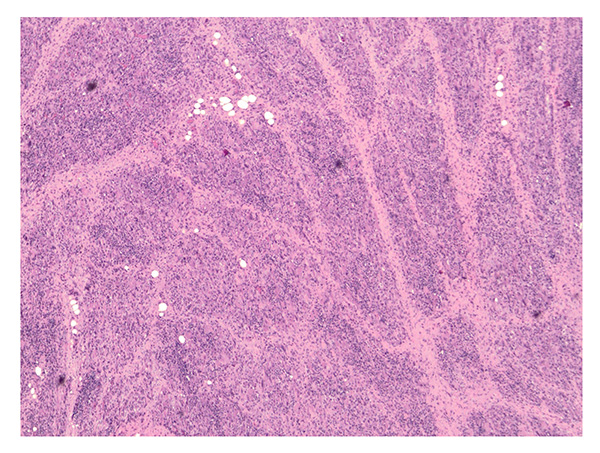
Figure 1: Hematoxylin and eosin staining shows muscle fascicles that are extensively obliterated by inflammatory infiltrates (40× magnification).
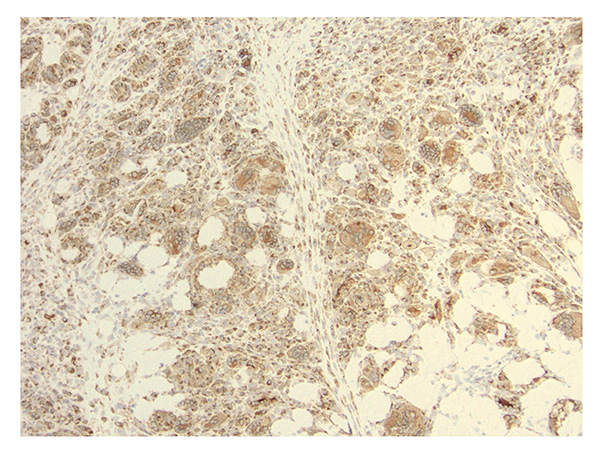
Figure 2: Immunohistochemical staining using 3,3′-diaminobenzidine (DAB) shows infiltrates including numerous CD68+ histiocytes/macrophages, labeled brown, including a few multinuclear giant cells (200× magnification).
After completion of 5 sessions of PLEX, the patient received intravenous immunoglobulin (IVIG) and pyridostigmine. She was continued on high-dose steroids. Over the next week, her respiratory status improved markedly, and she was able to be weaned from BiPAP to room air. After 2 weeks of hospitalization, creatine kinase level had normalized, proximal weakness had begun to improve, and she was discharged on azathioprine and a steroid taper.
After discharge, she developed nausea and vomiting that resolved with azathioprine discontinuation. The disease remained in remission for several months on IVIG, but as steroids were tapered weakness recurred, as did a moderate elevation of creatine kinase levels, prompting addition of methotrexate. Rituximab may be added, depending on her clinical response to methotrexate.
Discussion
IIM constitutes a group of systemic diseases characterized by skeletal muscle inflammation, progressive weakness, and a range of manifestations such as skin rash and lung involvement. Distinctive clinical phenotypes are associated with the presence of particular “myositis-specific” autoantibodies, although many patients with IIM are seronegative (as this patient was). MG is a disease of the neuromuscular junction resulting from autoantibodies targeting proteins of the postsynaptic membrane (classically, anti-acetylcholine receptor antibodies, as in this case), leading to fatigable weakness often involving the bulbar and/or respiratory muscles.
Concurrent IIM and MG is uncommon but has been reported in case series [1,2]; it most often occurs in the setting of a thymoma (not found in this patient) or in association with the use of immune checkpoint inhibitor therapy. Myocarditis may also be present [3]; fortunately, this patient showed no signs of cardiac involvement.
Histologic features of myositis occurring in the setting of MG vary, but a “giant cell myositis” subtype, characterized by histiocyte-rich inflammation and giant cells without granulomas, has been well-described [1,2,3]. This histologic subtype of myositis is quite rare and likely occurs with disproportionate frequency among those with concurrent MG. It may involve rapidly progressive weakness, respiratory failure, and high risk of death; in such cases, aggressive immunosuppression is mandatory. Treatments that have shown promise in case series include pyridostigmine, IVIG, and PLEX [1,2]. Successful treatment has commonly involved steroids, although their use (especially alone) may transiently worsen disease [2,4]. Azathioprine, methotrexate, and mycophenolate have been used successfully as oral steroid-sparing agents, and thymectomy may be beneficial even in the absence of a thymoma [1,5]. Additional agents, including rituximab, may be effective for refractory disease [5].
Authors
Jonathan Thaler, MD
Rheumatology Fellow
Hospital for Special Surgery

Kurenai Tanji, MD, PhD
Professor, Department of Pathology & Cell Biology
Columbia University
References
- Scangarello FA, Angel-Buitrago L, Lang-Orsini M, et al. Giant cell myositis associated with concurrent myasthenia gravis: a case-based review of the literature. Clin Rheumatol. 2021;40(9):3841-3851. doi: 10.1007/s10067-021-05619-5.
- Paik JJ, Corse AM, Mammen AL. The co-existence of myasthenia gravis in patients with myositis: a case series. Semin Arthritis Rheum. 2014;43(6):792-6. doi: 10.1016/j.semarthrit.2013.12.005.
- Oflazer P. Giant cell myositis and myocarditis revisited. Acta Myol. 2020;39(4):302-306. doi: 10.36185/2532-1900-033. PMID: 33458585; PMCID: PMC7783435.
- Miller RG, Milner-Brown HS, Mirka A. Prednisone-induced worsening of neuromuscular function in myasthenia gravis. Neurology. 1986;36(5):729-32. doi: 10.1212/wnl.36.5.729. PMID: 3703276.
- Narayanaswami P, Sanders DB, Wolfe G, et al. International consensus guidance for management of myasthenia gravis: 2020 update. Neurology. 2021 ;96(3):114-122. doi: 10.1212/WNL.0000000000011124.