Overlapping ANCA-Associated Vasculitis and IgG4-Related Disease in a 65-Year-Old Man with Acute Renal Failure
From Grand Rounds from HSS: Management of Complex Cases | Volume 11, Issue 3
Case Report
A 65-year-old man with a history of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV), congestive heart failure, and peripheral arterial disease presented with fatigue and diffuse pain. His AAV diagnosis dated back 15 years, when he developed constitutional symptoms, skin lesions, orchitis, heart failure, aortitis, scleritis, splenic infarction, and mononeuritis multiplex with high-titer myeloperoxidase (MPO) antibodies. He was treated with glucocorticoids, cyclophosphamide, and rituximab.
Upon his presentation to the emergency department, he had not had any treatment or monitoring for 10 years. He reported months of fatigue, abdominal pain, and diffuse musculoskeletal pain. Physical examination was significant only for subtle bibasilar crackles and diffuse joint tenderness without synovitis. Creatinine was elevated at 4.8 mg/dL; urinalysis showed 1.2 g of proteinuria and an active urine sediment. Other routine laboratory findings, including absolute eosinophil count, were normal. Hepatitis serologies were consistent with cleared hepatitis B virus infection. Serologies were notable for elevated perinuclear ANCA and MPO, total immunoglobulin G (IgG; 3,643 mg/dL), IgG1 (1,706 mg/dL), IgG4 (1,274 mg/dL), and IgE (305 mg/dL). Imaging of the abdomen and pelvis showed renal calcifications, splenomegaly, and retroperitoneal lymphadenopathy.
Renal biopsy demonstrated a crescentic glomerulonephritis (Fig. 1) consistent with a small vessel vasculitis. Histology also showed tissue eosinophils and a dense lymphoplasmacytic infiltrate (Fig. 2), patchy fibrosis throughout the interstitium, and tubular atrophy and injury. Immunohistochemistry showed strong positive staining for IgG in almost all the interstitial plasma cells, with nearly 50% of them staining positive for IgG4 (Fig. 3). The patient was diagnosed with overlapping microscopic polyangiitis (MPA) and IgG4-related disease (IgG4-RD). He improved with pulse steroids and rituximab and was discharged home with sustained renal recovery at follow-up visit 1 month later.
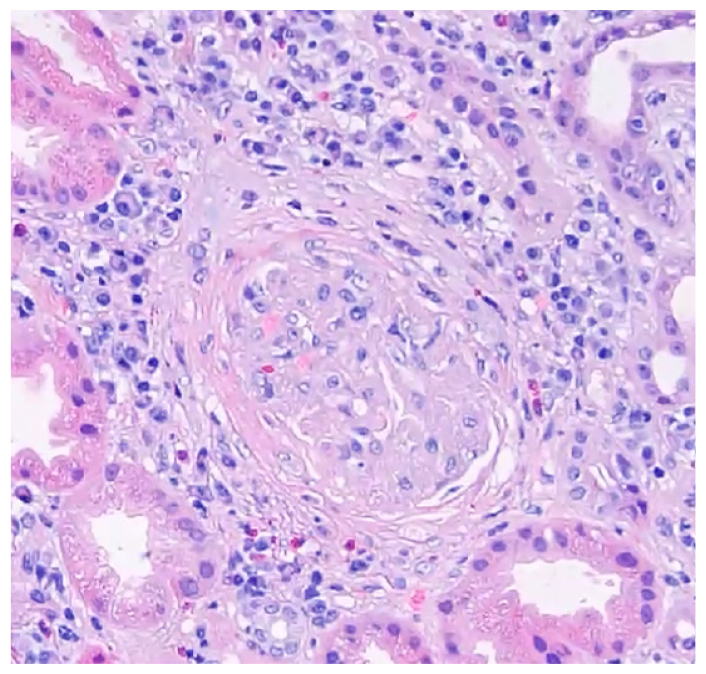
Figure 1: Hematoxylin and eosin (H&E) staining of renal biopsy shows fibrocellular crescent compressing the glomerulus.
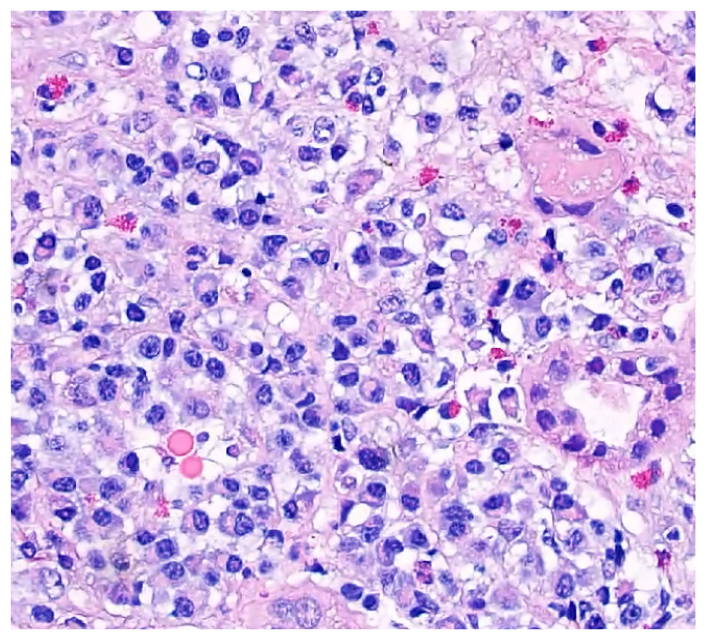
Figure 2: Renal histology shows lymphoplasmacytic infiltrate and tissue eosinophilia.
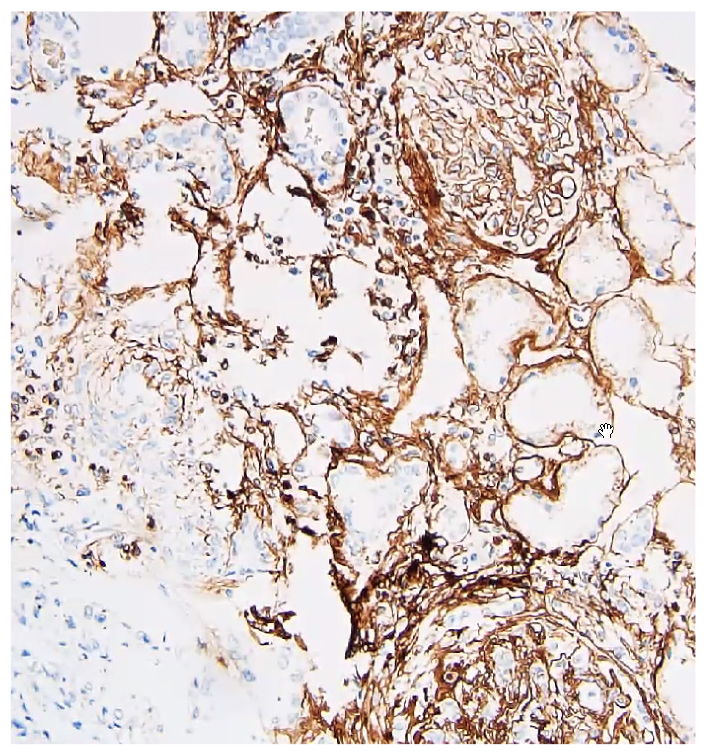
Figure 3: IgG4+ immunohistochemical staining.
Discussion
MPA is a systemic vasculitis that involves the kidneys in more than 90% of patients [1]. IgG4-RD is a systemic fibroinflammatory condition that can affect the kidneys with a tubulointerstitial nephritis or glomerulonephritis [2]. It appears that both disease processes contributed to this patient’s presentation. His history of small vessel (mononeuritis multiplex) and large vessel (aortitis) disease suggests he may have had overlapping disease all along.
Elevated serum IgG4+ cells can be seen in AAV and many other autoimmune conditions [3], yet this patient had several features indicating true IgG4-RD. He had marked lymphoplasmacytic tubulointerstitial infiltrates, which are more typical in IgG4-RD than in AAV. He had highly elevated serum IgG1, IgG4, and IgE levels and elevated IgG4+:IgG ratio on biopsy, all suggestive of systemic IgG4-RD [4]. Also, elevated IgG1 appears to be particularly associated with renal disease in those with systemic IgG4-RD [2].
Glucocorticoids and rituximab are the mainstay of treatment for both diseases when there is critical organ involvement [4,5]. This approach allows for targeting of both diseases with a single immunomodulatory regimen. Since both AAV and IgG4-RD are systemic inflammatory diseases potentially involving multiple systems, it is important to investigate for both diseases, particularly when presenting features are not classic of either syndrome.
Authors
Robert Spandorfer, MD
Rheumatology Fellow
Hospital for Special Surgery
References
- Almaani S, Fussner LA, Brodsky S, Meara AS, Jayne D. ANCA-associated vasculitis: an update. J Clin Med. 2021;10(7):1446. doi: 10.3390/ jcm10071446
- Martín-Nares E, Hernandez-Molina G, Rodríguez-Ramírez S, et al. IgG4-related kidney disease: experience from a Mexican cohort. Clin Rheumatol. 2020;39(11):3401–3408. doi: 10.1007/s10067- 020-05135-y
- Ebbo M, Grados A, Bernit E, et al. Pathologies associated with serum IgG4 elevation. Int J Rheumatol. 2012;2012:602809. doi: 10.1155/2012/602809
- Chung SA, Langford CA, Maz M, et al. 2021 American College of Rheumatology/Vasculitis Foundation guideline for the management of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol. 2021;73(8):1366– 1383. doi: 10.1002/art.41773
- Katz G, Stone JH. Clinical perspectives on IgG4-related disease and its classification. Annu Rev Med. 2022;73:545-562. doi: 10.1146/annurevmed- 050219-034449