Osteogenesis Imperfecta (Brittle Bone Disease)
What is osteogenesis imperfecta?
Osteogenesis imperfecta (OI), sometimes called “brittle bone disease," is a form of skeletal dysplasia. It is a congenital disorder in which a person is born with very weak bones, which leads to frequent fractures and other health problems.
What does osteogenesis imperfecta mean?
Osteogenesis imperfecta, from Latin and Greek roots, means “imperfect bone formation.” This genetic condition is characterized by fragile bones, fractures, skeletal deformities, short stature, and other symptoms.
What causes osteogenesis imperfecta?
Osteogenesis imperfecta results from an inability to produce a sufficient amount or normal quality of type I collagen. Type I collagen is the most common protein in bone and is responsible for maintaining bone strength.
The underlying genetic cause is one or more variations in any of various individual genes associated with the production or modification of this collagen, including COL1A1, COL1A2, CRTAP, P3H1 (also known as LEPRE1), and others.
The majority of cases of OI are caused by an autosomal dominant mutation in one of the genes responsible for producing or modifying type I collagen. Usually, when no one else in a patient’s family has a history of OI, the genetic variation is a spontaneous (“de novo”) mutation rather than an inherited trait.
Whether the cause is inherited or is de novo, genetic counseling for the parents is advised. However, in de novo cases, the chance of the parents having another child with the condition is small. Genetic counseling is also important for adults who have OI and wish to become parents. A person with osteogenesis imperfecta can have up to a 50% chance of having a child with OI in each pregnancy.
What are the symptoms of osteogenesis imperfecta?
The most common symptom is frequent bone fractures, although children with this condition may also have ligamentous laxity (loose ligaments), muscle weakness, brittle teeth, spinal deformities (scoliosis and/or kyphosis), and/or chronic pain issues. Short stature of varying severity is also a hallmark of OI.
Other related medical problems may also be present at birth, including respiratory difficulties resulting from rib cage deformities and underdevelopment of the lungs. Some people with OI may experience hearing loss (usually in their twenties), poor dentition, and/or blue sclera (the whites of the eyes being tinted somewhat blue, purple or gray).
What is the life expectancy in osteogenesis imperfecta?
Life expectancy in people with OI varies widely according to the severity of osteogenesis imperfecta. In the most common type, type I, patients can expect a normal life expectancy. In more severe forms of OI, life expectancy can be shortened.
What are the types of osteogenesis imperfecta?
There are many types of OI, with type I being the most common. Originally, four discrete types of OI were described. More recently, more uniquely described forms have been identified, although the four originally described types are the most common. The condition and prognosis of each affected patient varies, based on the type of OI and genetic mutation.
Osteogenesis imperfecta type I
This is the mildest and most common form. Some children may experience fractures in early infancy but, in others, this condition may remain undetected until latter childhood or adolescence, when fractures occur after a minor trauma or from no obvious cause. Children with OI type I may experience hearing loss, scoliosis, and weak teeth, but usually do not have severe short stature. Life expectancy is normal.
Osteogenesis imperfecta type II
This is the most severe form of the disease, often resulting in fractures in utero, as well as underdeveloped lungs. This may result in fetal death, or children born may die in early infancy as result of respiratory problems or other issues associated with severely impacted bones.
Osteogenesis imperfecta type III
This is also a severe form of OI. Children usually experience bone fractures in utero with continued fractures in early infancy/childhood. Significant bone deformities and short stature are expected. Children with this form may have respiratory issues, scoliosis, hearing loss, and weak teeth. Life expectancy is generally shorter than that of the general population.
Osteogenesis imperfecta type IV
Type IV is similar to type III and can range in severity but it is generally milder than type III. Children often have fractures in infancy/childhood. Children with OI type IV may have mild bone deformities and have shorter stature than the general population. Children with this form may also have scoliosis, hearing loss, and weak teeth.
What is the treatment for osteogenesis imperfecta?
Because children with this condition often have numerous medical needs, a number of specialists may be involved in their care and treatment, which may include nonsurgical drug therapies and/or surgery on a case-by-case basis. A multidisciplinary approach offers the best chance for reducing the number of fractures the child experiences, addressing skeletal deformities, maximizing function, and improving quality of life.
At HSS, this means that orthopedists work as a team with pediatricians, pediatric physical therapists, endocrinologists, genetic counselors, nurses, social workers/mental health workers, and nutritionists. Some of this care is delivered through the Kathryn O. and Alan C. Greenberg Center for Skeletal Dysplasias. Young patients sometimes also consult with a psychologist, since the frequent doctor visits, hospital stays, including surgeries, in early childhood can be emotionally distressing.
New advances in surgical technique and tools, combined with medication and physical therapy, are changing the course of this disease for many children.
Nonsurgical drug treatment for osteogenesis imperfecta
Although there is still no cure for osteogenesis imperfecta, many patients may derive significant protection against fractures from bisphosphonate drug therapy. It is also important that children with OI have adequate dietary calcium and vitamin D.
All individuals make osteoclasts (cells that remove or break down older bone) and osteoblasts (cells that build new bone) for a continual process of “bone remodeling.” This bone tissue replacement takes place throughout a child’s growth and, to a lesser extent, even after a person reaches skeletal maturity. Patients with more severe forms of OI tend to have very high turnover of bone remodeling, as well as malformed bone.
Bisphosphonates are medications originally developed for the treatment of osteoporosis and certain cancers. They slow down the rate at which osteoclasts take bone away, providing the patient with protective scaffolding, which is actually calcified cartilage.
In patients who are appropriate candidates, bisphosphonate therapy should be initiated as early as possible. Children as young as one to two months with severe forms of OI can receive intravenous (IV) infusions of bisphosphonates every few weeks in a hospital setting. As the child becomes older, the medication may be administered on an outpatient basis or on an inpatient basis at a less frequent interval. In children with milder forms of OI, the decision on when to begin this medication depends on the incidence of fractures and our multidisciplinary team.
While most children tolerate these medications very well, at HSS, we follow them carefully with the help of a pediatric endocrinologist (a physician who specializes in hormonal and metabolic disorders in children). The child’s blood is tested regularly to monitor calcium, phosphorous, and vitamin D levels. DEXA scans (a type of X-ray) which assess bone quality and bone mass are administered yearly, after age three to four.
Bisphosphonate therapy requires an individualized approach to each patient. It is important to monitor the long-term effects on the patient’s bones.
Bisphosphonates have been shown to interfere with the normal healing process after a surgical osteotomy and therefore, bisphosphonate therapy may need to be held prior to and after surgical interventions until it is established that the bone is healed.
Surgery for osteogenesis imperfecta – fixation and nailing
Although orthopedic intervention for osteogenesis imperfecta can include bracing to help protect the patient during physical activity and physical therapy, surgery with intramedullary (in-bone) fixation to correct deformity offers the greatest long-term benefit and protection against future fracture.
Intramedullary fixation
Historically, orthopedists used rods of a fixed length to help fractures heal and avoid or correct deformity. However, as the child grew, he or she was at risk of fracturing the leg immediately below the rod, because the bone would grow while the rod would not. Today, when appropriate, the pediatric orthopedists at HSS use the Fassier-Duval Telescopic Intramedullary System. This is a form of instrumentation designed specifically for patients with OI who are still growing.
Fassier-Duval rods are secured on the far ends of the bone at each growth plate and telescope, or extend, as growth in the bone occurs. Following the initial placement, there is less need for surgical adjustment while the child grows compared to a non-telescoping, or static, rod. The patient is therefore less likely to develop the type of fractures associated with older, non-telescoping rods, and may require fewer surgeries as well.
X-rays depicting progression of the Fassier-Duval telescopic intramedullary system
This series of X-rays depicts the surgical progression that addresses the needs of a growing child with OI.
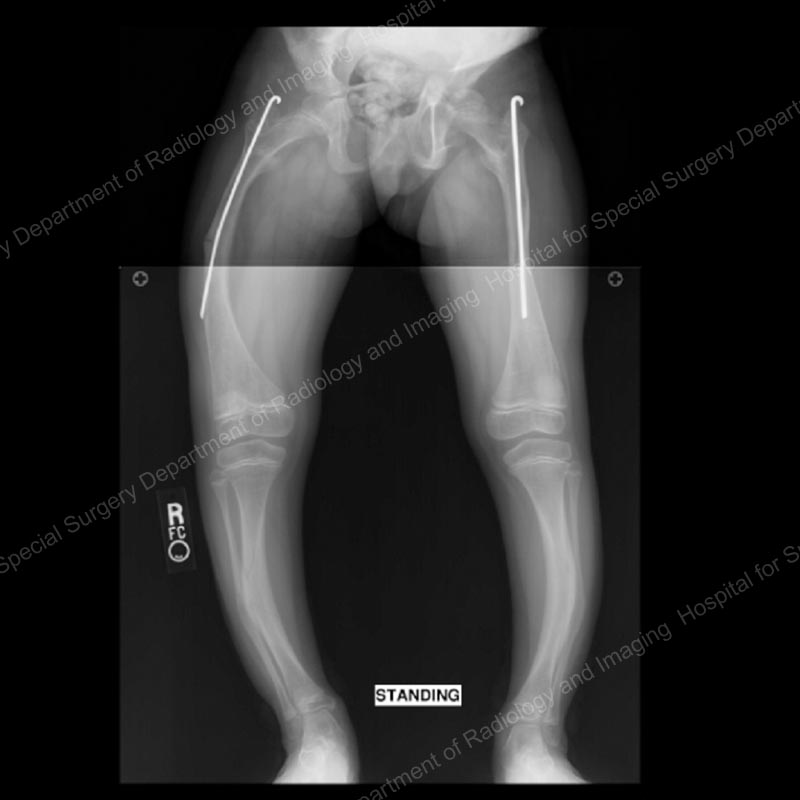
Figure 1: Anteroposterior (front-to-back) X-ray of the patient when she was initially treated with static intramedullary rods. As she outgrew these rods, they were replaced with Fassier-Duval rods (Figures 2 through 4).
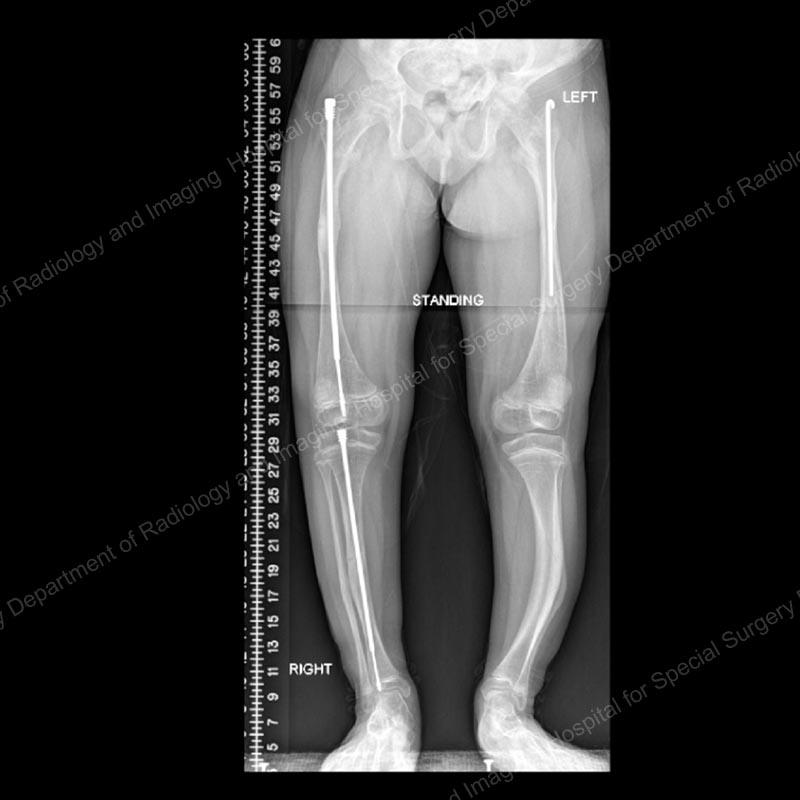
Figure 2: Anteroposterior (front-to-back) X-ray after osteotomies and rod placement in the femur and tibia of the right leg. This demonstrates how Fassier-Duval rods provide stability for the fragile bones, allow for correction of bowing, and lengthen as the child grows.
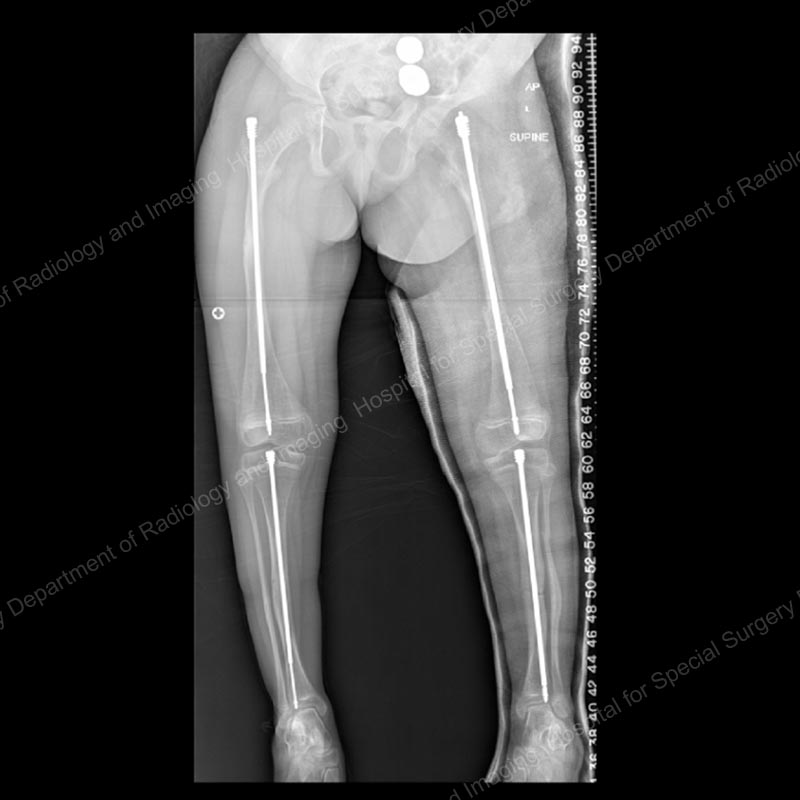
Figure 3: Posteroanterior (back-to-front) X-ray showing continued lengthening of the right femur and tibia rods, as well as placement of Fassier-Duval rods in the left femur and tibia.
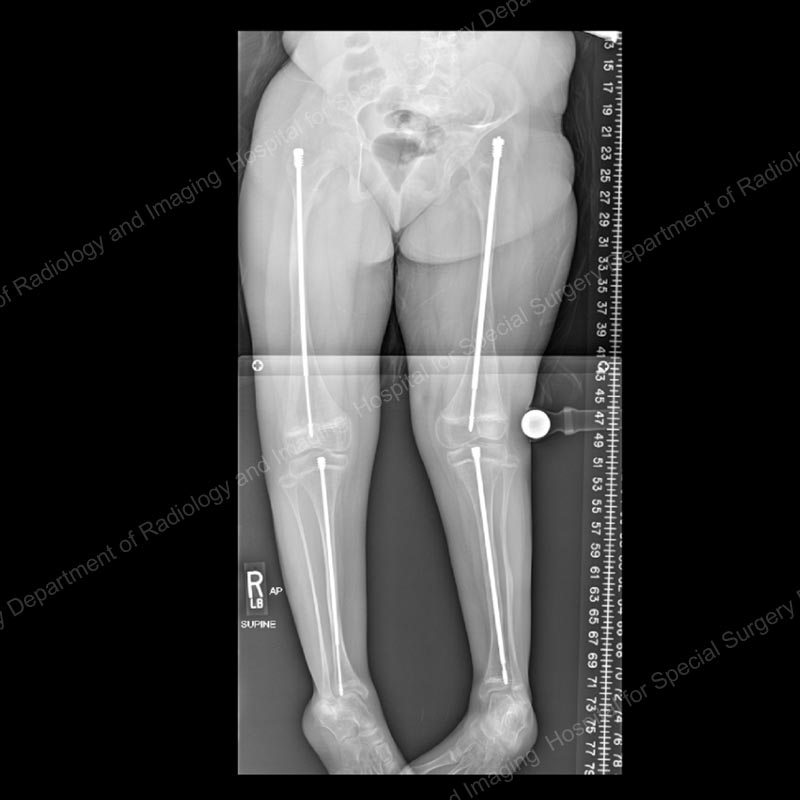
Figure 4: Posteroanterior (back-to-front) X-ray showing continued lengthening of the rods.
Physical therapy is an especially important component of care following rodding surgery or any other procedure used to treat of fractures. Muscle mass decreases in any person who is immobilized, so post-surgical therapy can be intensive. We see the patients two or three times a week and give them safe exercises to regain their strength.
Physical therapy and prognosis for osteogenesis imperfecta patients
Regardless of the type of treatment they receive, physical therapy is necessary to safely navigate daily routines. Maintaining or improving muscle and bone strength are goals for all children with osteogenesis imperfecta. Physical therapists also work closely with families of patients, especially to teach parents how safely carry, dress, change diapers and otherwise handle their infants in ways that protects them from fractures.
As babies become toddlers and young children, they must also be taught how to move safely, avoiding jarring movements, jerking, and twisting. As patients reach adolescence, there tends to be a reduction in fractures. Teenagers and college-bound patients decide whether they want to continue with physical therapy. Some older children and young adults opt to continue walking, while others find they can participate more easily in school and other activities by using a wheelchair.
Into adulthood, some OI patients experience pain, fatigue, sleep disturbances, as well as respiratory and gastrointestinal issues. A patient’s care team may expand to include psychologists, if it has not already done so, to help manage any psychological affects these symptoms may cause. It is important that adults with OI continue to follow with a multidisciplinary team.
Related articles
- Skeletal Dysplasias
- Wheelchair-Based Exercises for People with Osteogenesis Imperfecta
- Low Bone Density and Osteoporosis in Children
- Broken Elbows in Children: An Overview of Elbow Fractures
- Tibial Spine Fractures in Children: What to Know
- Femur Fractures in Children: Treating a Child’s Broken Thighbone
Osteogenesis Imperfecta (Brittle Bone Disease) Patient Stories
In the news
Updated: 8/26/2024
Reviewed and updated by Daniel W. Green, MD, MS, FAAP; FACS Jessica H. Heyer, MD;
Cathleen L. Raggio, MD; Alyson Weiner, MD
References
- Bronheim R, Khan S, Carter E, Sandhaus RA, Raggio C. Scoliosis and Cardiopulmonary Outcomes in Osteogenesis Imperfecta Patients. Spine (Phila Pa 1976). 2019 Aug 1;44(15):1057-1063. doi: 10.1097/BRS.0000000000003012. PMID: 31335789. https://pubmed.ncbi.nlm.nih.gov/31335789/
- Hald JD, Langdahl B, Folkestad L, Wekre LL, Johnson R, Nagamani SCS, Raggio C, Ralston SH, Semler O, Tosi L, Orwoll E. Osteogenesis Imperfecta: Skeletal and Non-skeletal Challenges in Adulthood. Calcif Tissue Int. 2024 Jun 5. doi: 10.1007/s00223-024-01236-x. Epub ahead of print. PMID: 38836890. https://pubmed.ncbi.nlm.nih.gov/38836890/
- Hidalgo Perea S, Green DW. Osteogenesis imperfecta: treatment and surgical management. Curr Opin Pediatr. 2021 Feb 1;33(1):74-78. doi: 10.1097/MOP.0000000000000968. PMID: 33278111. https://pubmed.ncbi.nlm.nih.gov/33278111/
- Westerheim I, Hart T, van Welzenis T, Wekre LL, Semler O, Raggio C, Bober MB, Rapoport M, Prince S, Rauch F. The IMPACT survey: a mixed methods study to understand the experience of children, adolescents and adults with osteogenesis imperfecta and their caregivers. Orphanet J Rare Dis. 2024 Mar 21;19(1):128. doi: 10.1186/s13023-024-03126-9. PMID: 38515144; PMCID: PMC10956293. https://pubmed.ncbi.nlm.nih.gov/38515144/
- Yonko EA, Emanuel JS, Carter EM, Sandhaus RA, Raggio CL. Respiratory impairment impacts QOL in osteogenesis imperfecta independent of skeletal abnormalities. Arch Osteoporos. 2020 Oct 2;15(1):153. doi: 10.1007/s11657-020-00818-0. PMID: 33009598. https://pubmed.ncbi.nlm.nih.gov/33009598/